
AlphaFold 3:蛋白质结构预测的革命性进展
AlphaFold 3是人工智能在生物学领域的一项重大突破,它为准确预测生物分子的结构和相互作用开辟了新的可能。本文将深入探讨AlphaFold 3的PyTorch实现,解析其核心技术和创新点,为研究者和开发者提供宝贵的参考。
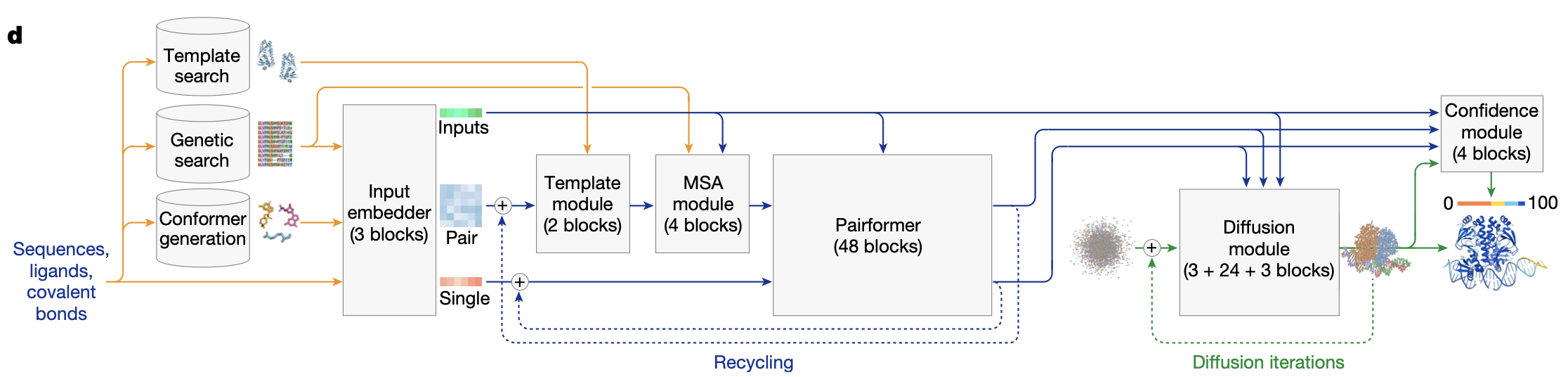
AlphaFold 3的核心创新
AlphaFold 3相比于前代模型有以下几个关键的创新:
-
更广泛的应用范围:不仅可以预测蛋白质结构,还能预测核酸、配体等多种生物分子的结构和相互作用。
-
改进的网络架构:引入了PairFormer模块替代了EvoFormer,能更有效地处理配对表示和单独表示。
-
扩散模型的应用:直接在原子坐标上操作,通过去噪任务学习蛋白质结构的多尺度特征。
-
交叉蒸馏技术:通过enriching训练数据来抑制模型的幻觉问题。
-
置信度预测:能够预测原子层面和配对层面的误差,提高结果的可靠性。
这些创新使AlphaFold 3在准确性和适用范围上都有了显著提升。
PyTorch实现的关键模块
在PyTorch的实现中,主要包含以下几个核心模块:
-
输入嵌入模块:处理序列、MSA和模板信息。
-
PairFormer模块:替代了EvoFormer,处理配对表示和单独表示。
-
扩散模块:直接在原子坐标上进行操作。
-
置信度预测模块:预测结构的误差。
下面是一个简化的使用示例:
import torch
from alphafold3_pytorch import Alphafold3
# 初始化模型
alphafold3 = Alphafold3(
dim_atom_inputs = 77,
dim_template_feats = 108
)
# 准备输入数据
seq_len = 16
atom_inputs = torch.randn(2, seq_len, 77)
atompair_inputs = torch.randn(2, seq_len, seq_len, 5)
# ... 其他输入 ...
# 训练
loss = alphafold3(
num_recycling_steps = 2,
atom_inputs = atom_inputs,
atompair_inputs = atompair_inputs,
# ... 其他参数 ...
)
loss.backward()
# 推理
sampled_atom_pos = alphafold3(
num_recycling_steps = 4,
num_sample_steps = 16,
atom_inputs = atom_inputs,
atompair_inputs = atompair_inputs,
# ... 其他参数 ...
)
数据准备和处理
AlphaFold 3的训练需要大量高质量的数据。主要的数据来源是蛋白质数据库(PDB)。数据处理流程包括:
- 下载PDB数据
- 数据过滤
- 数据聚类
这个过程确保了训练数据的质量和多样性。详细的数据处理脚本和步骤可以在项目的GitHub仓库中找到。
模型训练和推理
AlphaFold 3的训练过程涉及多个损失函数,包括距离图预测、原子位置预测等。训练时需要大量的计算资源,通常需要使用GPU集群。
在推理阶段,模型会生成多个样本,然后根据置信度选择最佳结果。这种方法能够提高预测的稳定性和准确性。
社区贡献和未来展望
AlphaFold 3的开源实现得到了广泛的社区支持。许多研究者和开发者为项目贡献了宝贵的改进和功能扩展。这种开放合作的模式极大地推动了项目的发展。
未来,AlphaFold 3还有很大的发展空间:
- 进一步提高对大型复合物的预测能力
- 改进对动态结构的预测
- 扩展到更多类型的生物分子
- 提高计算效率,使其能在更普及的硬件上运行
结论
AlphaFold 3代表了蛋白质结构预测领域的一个重要里程碑。它的PyTorch实现为研究者和开发者提供了一个强大的工具,有望加速生物学和药物研发等领域的创新。随着社区的不断贡献和技术的持续进步,我们可以期待看到更多令人兴奋的应用和突破。
参考资源
通过深入了解AlphaFold 3及其PyTorch实现,研究者和开发者可以更好地利用这一强大工具,推动生命科学研究的边界。让我们共同期待AlphaFold 3带来的更多突破性进展!









