
Sarek简介
Sarek是一款由nf-core社区开发的高通量生物信息学分析流程,专门用于从全基因组测序(WGS)或靶向测序数据中检测基因组变异。该流程最初针对人类和小鼠样本设计,但实际上可以应用于任何具有参考基因组的物种。Sarek不仅可以处理单个样本,还支持肿瘤/正常配对样本的分析,甚至可以包含额外的复发样本。
Sarek流程基于Nextflow工作流管理系统构建,具有极强的可移植性,可以在多种计算基础设施上运行。它采用Docker/Singularity容器技术,使得软件安装变得简单,同时保证了结果的高度可重复性。Sarek实现了Nextflow DSL2,每个处理步骤使用单独的容器,这使得维护和更新软件依赖变得更加容易。大多数处理模块都已提交并安装到nf-core/modules仓库中,以便所有nf-core流程和Nextflow社区成员可以使用。
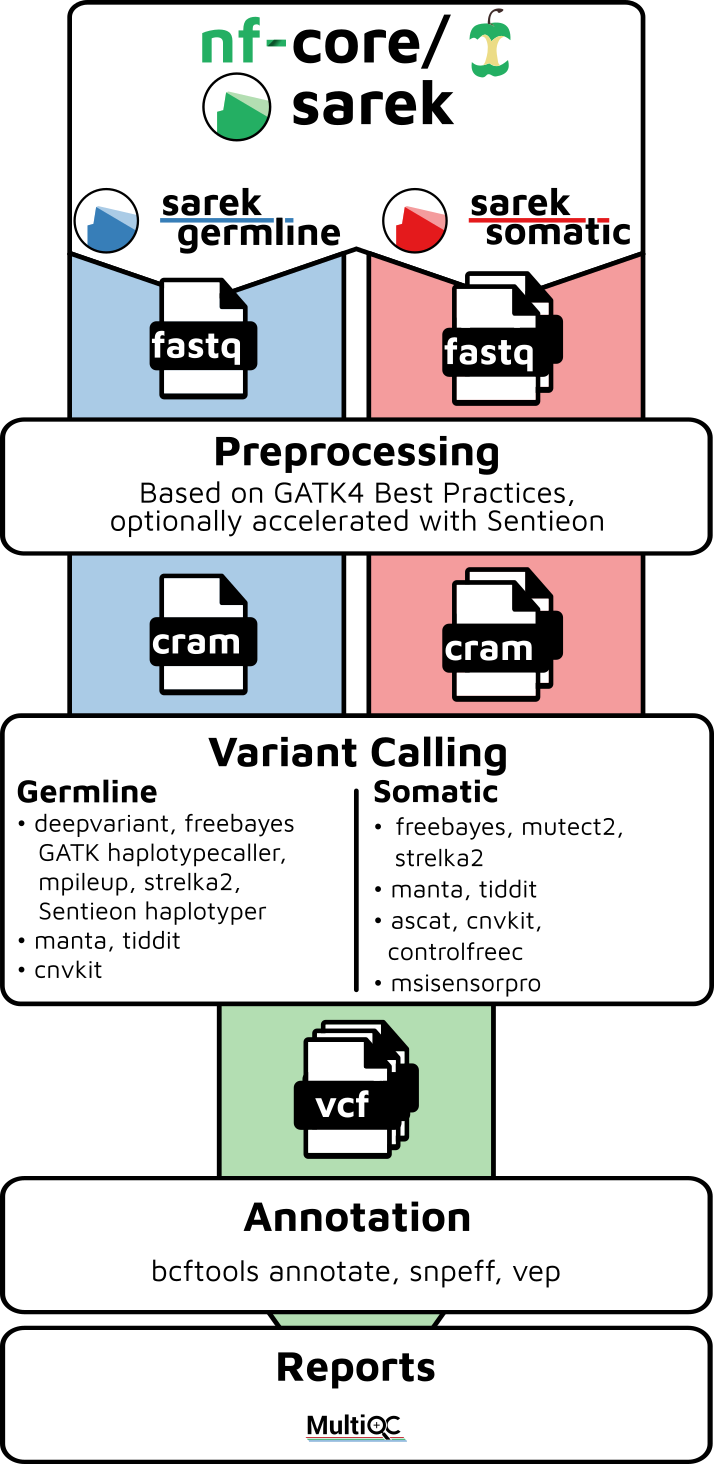
Sarek的主要功能
Sarek流程根据提供的选项和样本,可以执行以下主要功能:
- 从UMI序列形成一致性读段(使用fgbio)
- 测序质量控制和修剪(使用FastQC和fastp,通过--trim_fastq选项启用)
- 将读段比对到参考基因组(可选择BWA-mem, BWA-mem2, dragmap或Sentieon BWA-mem)
- 处理BAM文件(使用GATK MarkDuplicates, GATK BaseRecalibrator和GATK ApplyBQSR,或Sentieon LocusCollector和Sentieon Dedup)
- 总结比对统计信息(使用samtools stats和mosdepth)
- 变异检测(通过--tools选项启用,支持多种工具如ASCAT, CNVkit, Control-FREEC, DeepVariant, freebayes, GATK HaplotypeCaller等)
- 变异过滤和注释(使用SnpEff, Ensembl VEP, BCFtools annotate)
- 汇总和展示质量控制结果(使用MultiQC)
Sarek的工作流程可以形象地用"地铁线路图"来表示:
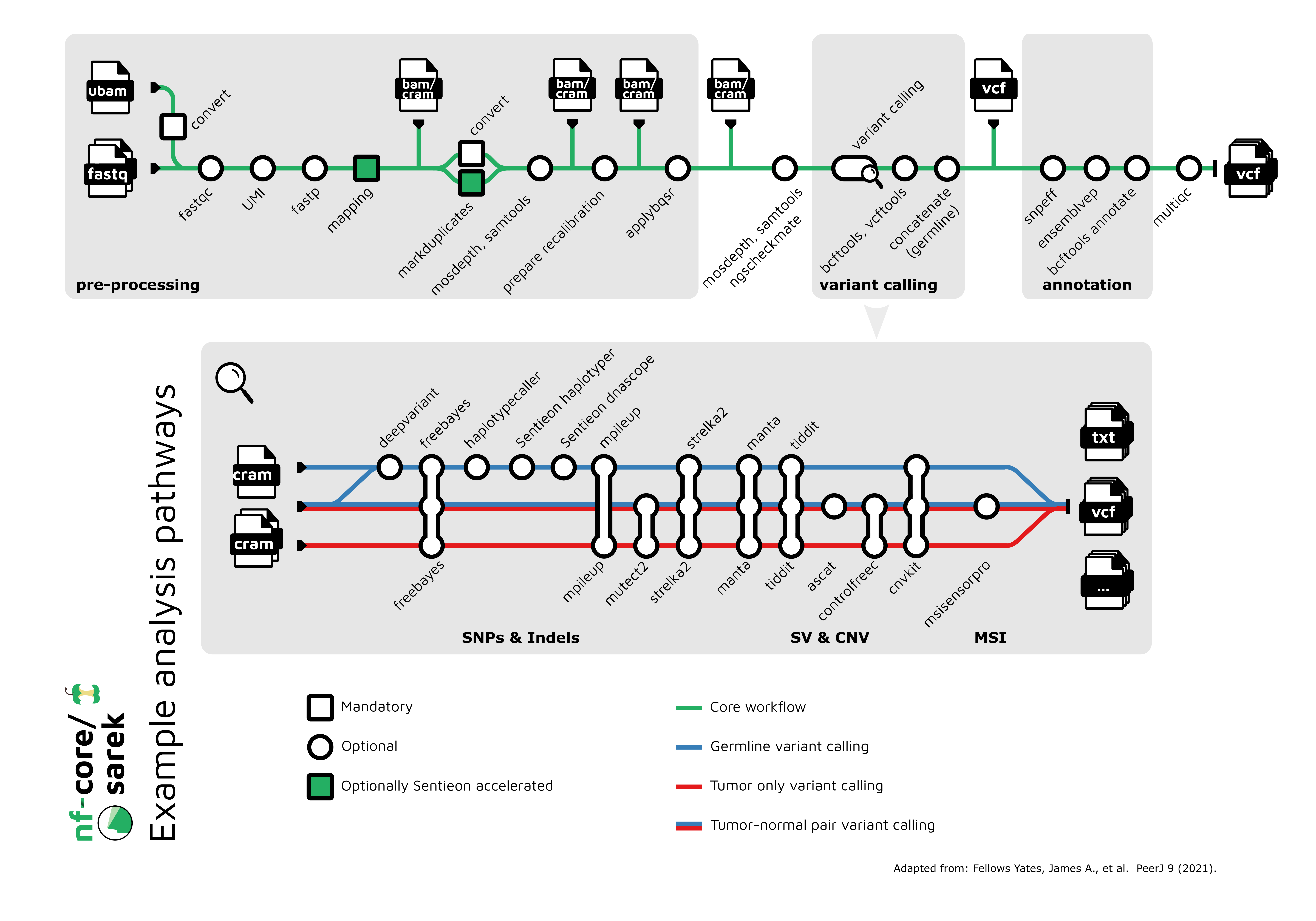
这个"地铁线路图"清晰地展示了Sarek流程中各个分析步骤的逻辑关系和数据流向,帮助用户直观地理解整个分析过程。
如何使用Sarek
要使用Sarek,首先需要准备一个包含输入数据信息的样本表。样本表是一个CSV格式的文件,每一行代表一对fastq文件(双端测序)。例如:
patient,sample,lane,fastq_1,fastq_2
ID1,S1,L002,ID1_S1_L002_R1_001.fastq.gz,ID1_S1_L002_R2_001.fastq.gz
准备好样本表后,可以使用以下命令运行Sarek流程:
nextflow run nf-core/sarek \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>
需要注意的是,流程参数应该通过命令行界面(CLI)或Nextflow的-params-file选项提供。自定义配置文件(包括通过Nextflow的-c选项提供的文件)可以用于提供除参数之外的任何配置。
对于更详细的使用说明和更多功能,请参考Sarek使用文档和参数文档。
Sarek的输出结果
Sarek流程的输出结果包括多个方面,涵盖了从原始数据处理到最终变异检测和注释的各个环节。为了了解Sarek在全尺寸数据集上的表现,可以查看nf-core网站上的结果页面。这里展示了使用完整数据集运行测试的结果,可以作为benchmark参考。
关于输出文件和报告的更多详细信息,请参考Sarek输出文档。
Sarek的性能评估
为了评估Sarek的性能和可靠性,开发团队在每次发布新版本时都会进行三组全尺寸测试:
test_full: 使用来自SEQ2C联盟的一个患者的肿瘤-正常配对数据test_full_germline: 使用一个30X覆盖度的WGS Genome-in-a-Bottle(NA12878)数据集test_full_germline_ncbench_agilent: 使用两个WES样本,分别有7500万和2亿个读段
这些测试的结果会上传到Zenodo,并与真实数据集进行比较评估。评估结果可以通过NCBench仪表板查看。这种持续的性能评估确保了Sarek流程的稳定性和可靠性。
Sarek的开发团队和贡献者
Sarek最初由Maxime U Garcia和Szilveszter Juhos在瑞典国家基因组基础设施(NGI)和瑞典国家生物信息学基础设施(NBIS)开发,得到了瑞典儿童肿瘤生物银行(Barntumörbanken)的支持。后来,来自QBiC的Friederike Hanssen和Gisela Gabernet加入并协助了进一步的开发。
Sarek流程向Nextflow DSL2的转换主要由Friederike Hanssen和Maxime U Garcia领导完成。目前,流程的维护工作主要由Friederike Hanssen和Maxime U Garcia(现就职于Seqera Labs)负责。
除了核心开发团队,还有许多贡献者为Sarek的发展做出了重要贡献。这些贡献者来自世界各地的研究机构和企业,他们的专业知识和努力使Sarek成为一个强大而灵活的生物信息学工具。
如何为Sarek做出贡献
Sarek是一个开源项目,欢迎社区成员参与贡献。如果您想为Sarek做出贡献,可以查看贡献指南。无论是报告问题、提出新功能建议,还是直接提交代码,都是对项目很有价值的贡献。
如果您需要进一步的信息或帮助,可以加入Slack上的#sarek频道(可以通过这个邀请链接加入)。您也可以直接联系项目的主要维护者:Maxime U Garcia或Friederike Hanssen。
Sarek的引用
如果您在研究中使用了Sarek,请引用以下文章:
Friederike Hanssen, Maxime U Garcia, Lasse Folkersen, Anders Sune Pedersen, Francesco Lescai, Susanne Jodoin, Edmund Miller, Oskar Wacker, Nicholas Smith, nf-core community, Gisela Gabernet, Sven Nahnsen Scalable and efficient DNA sequencing analysis on different compute infrastructures aiding variant discovery NAR Genomics and Bioinformatics Volume 6, Issue 2, June 2024, lqae031, doi: 10.1093/nargab/lqae031.
Garcia M, Juhos S, Larsson M et al. Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants [version 2; peer review: 2 approved] F1000Research 2020, 9:63 doi: 10.12688/f1000research.16665.2.
您还可以引用Sarek的Zenodo记录,使用DOI:10.5281/zenodo.3476425。
此外,Sarek使用了许多其他工具和软件包。这些工具的详细引用信息可以在CITATIONS.md文件中找到。建议在发表研究结果时,同时引用这些工具,以表彰它们对您研究的贡献。
结语
Sarek作为一个功能强大、灵活且易于使用的生物信息学流程,为基因组变异分析提供了一个全面的解决方案。它不仅整合了最新的生物信息学工具和技术,还通过严格的性能评估和持续的社区贡献不断完善。无论您是进行基础研究、临床诊断还是大规模基因组学项目,Sarek都能满足您的需求,帮助您高效、准确地完成基因组变异分析任务。
随着生物信息学领域的快速发展,Sarek也在不断更新和改进。我们鼓励用户关注项目的GitHub仓库以获取最新版本和更新信息。同时,我们也欢迎更多的研究者和开发者加入Sarek社区,共同推动这个强大工具的发展,为生命科学研究做出贡献。









