
 访问官网
访问官网 Github
Github 文档
文档 论文
论文

用于免疫组织化学图像定量的深度学习推断多重免疫荧光
Nature MI'22 | CVPR'22 | MICCAI'23 | Histopathology'23 | MICCAI'24 | 云端部署 | 文档 | 支持
常规免疫组织化学(IHC)染色评估的生物标志物报告在诊断病理实验室中被广泛用于患者护理。目前,临床报告主要是定性或半定量的。通过创建一个称为DeepLIIF的多任务深度学习框架,我们提供了一个单步解决方案,用于染色脱色/分离、细胞分割和定量单细胞IHC评分。利用同一切片的IHC和多重免疫荧光(mpIF)染色共注册的独特新数据集,我们对低成本且普遍的IHC切片进行分割并转换为更昂贵但信息丰富的mpIF图像,同时为叠加的明场IHC通道提供必要的真实标准。此外,我们引入了一种新的细胞核包膜染色LAP2beta,其细胞覆盖率高(>95%),以改善IHC切片上的细胞描绘/分割和蛋白质表达定量。通过同时将输入的IHC图像转换为清晰/分离的mpIF通道并执行细胞分割/分类,我们展示了我们在清晰的IHC Ki67数据上训练的模型可以推广到更加嘈杂和有伪影的图像,以及其他核和非核标记物,如CD3、CD8、BCL2、BCL6、MYC、MUM1、CD10和TP53。我们在公开可用的基准数据集上以及与病理学家的半定量评分进行了全面评估。在IHC上训练的DeepLIIF可以很好地推广到H&E图像,实现开箱即用的细胞核分割。
DeepLIIF作为免费公开可用的云原生平台(https://deepliif.org)部署,支持Bioformats(支持150多种输入格式)和MLOps流程。我们还发布了**DeepLIIF**的单/多GPU训练、Torchserve/Dask+Torchscript部署和通过Pulumi实现自动扩展(支持1000多个并发连接)的实现;详细信息可以在我们的[文档](https://nadeemlab.github.io/DeepLIIF/)中找到。**DeepLIIF**可以通过[pip安装包](https://github.com/nadeemlab/DeepLIIF/edit/main/README.md#installing-deepliif)并使用deepliif CLI命令在本地运行(需要GPU)。DeepLIIF可以通过https://deepliif.org 网站、通过Python调用云API或通过ImageJ/Fiji插件远程使用(无需GPU);免费云原生平台的详细信息可以在我们的CVPR'22论文中找到。
© 本代码仅供非商业学术用途使用。

 DeepLIIF流程概述和样本输入IHC(不同的棕色/DAB标记物——BCL2、BCL6、CD10、CD3/CD8、Ki67)及其对应的DeepLIIF生成的苏木精/mpIF模态和分类(阳性(红色)和阴性(蓝色)细胞)分割掩码。(a) DeepLIIF概述。给定IHC输入,我们的多任务深度学习框架同时推断出相应的苏木精通道、mpIF DAPI、mpIF蛋白表达(Ki67、CD3、CD8等)和阳性/阴性蛋白细胞分割,将可解释性和可解释性直接嵌入模型本身,而不是依赖于粗略的激活/注意力图。在分割掩码中,红色细胞表示蛋白表达阳性的细胞(输入IHC中的棕色/DAB细胞),而蓝色细胞代表阴性细胞(输入IHC中的蓝色细胞)。(b) 不同IHC标记物的DeepLIIF生成的苏木精/mpIF模态和分割掩码示例。DeepLIIF经过清晰的IHC Ki67核标记物图像训练后,可以推广到更嘈杂的以及其他IHC核/细胞质标记物图像。
DeepLIIF流程概述和样本输入IHC(不同的棕色/DAB标记物——BCL2、BCL6、CD10、CD3/CD8、Ki67)及其对应的DeepLIIF生成的苏木精/mpIF模态和分类(阳性(红色)和阴性(蓝色)细胞)分割掩码。(a) DeepLIIF概述。给定IHC输入,我们的多任务深度学习框架同时推断出相应的苏木精通道、mpIF DAPI、mpIF蛋白表达(Ki67、CD3、CD8等)和阳性/阴性蛋白细胞分割,将可解释性和可解释性直接嵌入模型本身,而不是依赖于粗略的激活/注意力图。在分割掩码中,红色细胞表示蛋白表达阳性的细胞(输入IHC中的棕色/DAB细胞),而蓝色细胞代表阴性细胞(输入IHC中的蓝色细胞)。(b) 不同IHC标记物的DeepLIIF生成的苏木精/mpIF模态和分割掩码示例。DeepLIIF经过清晰的IHC Ki67核标记物图像训练后,可以推广到更嘈杂的以及其他IHC核/细胞质标记物图像。
先决条件
- Python 3.8
- Docker
安装deepliif
可以通过pip安装DeepLIIF:
$ conda create --name deepliif_env python=3.8
$ conda activate deepliif_env
(deepliif_env) $ conda install -c conda-forge openjdk
(deepliif_env) $ pip install deepliif
该包由两部分组成:
- 一个实现用于训练和测试DeepLIIF模型的核心功能的库。
- 一个CLI,用于运行常见的批处理操作,包括训练、批量测试和Torchscipt模型序列化。
你可以列出所有可用的命令:
(venv) $ deepliif --help
用法:deepliif [选项] 命令 [参数]...
选项:
--help 显示此消息并退出
命令:
prepare-testing-data 准备测试数据
serialize 使用Torchscript序列化DeepLIIF模型
test 测试已训练的模型
train 用于多任务图像转换模型的通用训练脚本
注意: 你可能需要安装与你的CUDA版本兼容的PyTorch版本。
否则,只会使用CPU。
访问PyTorch网站了解详情。
你可以通过检查以下代码是否返回True来确认你的安装是否能在GPU上运行:
import torch
torch.cuda.is_available()
训练数据集
对于训练,所有图像集必须为512x512,并组合成3072x512的图像(六张512x512大小的图像水平拼接在一起)。 数据需要按以下顺序排列:
XXX_Dataset
├── train
└── val
我们在CLI中提供了一个简单的函数来准备训练数据。
- 准备训练数据,你需要在输入目录中有每个图像的图像数据集(包括IHC、苏木精通道、mpIF DAPI、mpIF Lap2、mpIF标记物和分割掩码)。
单个图像集的六张图像必须具有相同的命名格式,只有表示图像类型的标签名称在它们之间有所不同。标签名称必须分别为:IHC、Hematoxylin、DAPI、Lap2、Marker、Seg。
该命令需要包含图像集数据的目录地址和输出数据集目录的地址。
它首先在给定的输出数据集目录中创建train和validation目录。
然后读取输入目录中的所有图像,并根据给定的
validation_ratio将组合后的图像保存在train或validation目录中。
deepliif prepare-training-data --input-dir /path/to/input/images
--output-dir /path/to/output/images
--validation-ratio 0.2
训练
要训练模型:
deepliif train --dataroot /path/to/input/images
--name Model_Name
或
python train.py --dataroot /path/to/input/images
--name Model_Name
- 要查看训练损失和结果,请打开URL http://localhost:8097。对于云服务器,请将localhost替换为你的IP。
- 每个epoch的中间训练结果位于
DeepLIIF/checkpoints/Model_Name/web/index.html。 - 训练好的模型默认将保存在
DeepLIIF/checkpoints/Model_Name中。 - 训练数据集可以在这里下载。
DP:要训练模型,你可以使用DP。DP是单进程的。这意味着你想使用的所有GPU必须在同一台机器上,这样它们才能包含在同一个进程中 - 除非你编写自己的代码来处理节点间(节点=机器)通信,否则你无法在多个GPU机器之间分配训练。 为了在同一进程中分割和管理多个GPU的工作负载,DP使用多线程。 你可以在这里找到更多关于DP的信息。
使用DP训练模型(以2个GPU(在1台机器上)为例):
deepliif train --dataroot <data_dir> --batch-size 6 --gpu-ids 0 --gpu-ids 1
请注意,batch-size是按每个进程定义的。由于DP是单进程方法,你设置的batch-size就是实际的批量大小。
DDP:要训练模型,可以使用DDP。DDP通常会启动多个进程。
DeepLIIF的代码遵循PyTorch的建议,每个GPU分配1个进程(文档)。如果你想为每个进程分配多个GPU,你需要修改DeepLIIF的代码(参见文档)。
尽管DDP有诸多优点,但一个缺点是需要额外的GPU内存用于通信的专用CUDA缓冲区。这里有一个简短的讨论链接。在DeepLIIF的背景下,这意味着可能会出现使用DP时可以使用更大的批量大小的情况,相比之下,DDP使用较小的批量大小可能实际上训练得更快。
你可以在这里找到更多关于DDP的信息。
要在本地机器上使用DDP启动训练,使用deepliif trainlaunch。以2个GPU(在1台机器上)为例:
deepliif trainlaunch --dataroot <数据目录> --batch-size 3 --gpu-ids 0 --gpu-ids 1 --use-torchrun "--nproc_per_node 2"
注意
batch-size是针对每个进程定义的。由于DDP是单进程方法,你设置的batch-size是每个进程的批量大小,实际批量大小将是batch-size乘以你启动的进程数。在上面的例子中,是3 * 2 = 6。- 你仍需要向训练命令提供所有要使用的GPU id。在内部,DeepLIIF在每个进程中使用
gpu_ids[local_rank]选择设备。如果你提供--gpu-ids 2 --gpu-ids 3,本地排名为0的进程将使用gpu id 2,本地排名为1的进程将使用gpu id 3。 -t 3 --log_dir <日志目录>不是必需的,但在torchrun中是一个有用的设置,它将每个进程的日志保存到你指定的日志目录。例如:
deepliif trainlaunch --dataroot <数据目录> --batch-size 3 --gpu-ids 0 --gpu-ids 1 --use-torchrun "-t 3 --log_dir <日志目录> --nproc_per_node 2"
- 如果你的PyTorch版本低于1.10,DeepLIIF在后端调用
torch.distributed.launch。否则,DeepLIIF调用torchrun。
序列化模型
安装的deepliif使用Dask对输入的IHC图像进行推理。
在运行test命令之前,必须使用Torchscript序列化模型文件。
要序列化模型文件:
deepliif serialize --model-dir /path/to/input/model/files
--output-dir /path/to/output/model/files
- 默认情况下,模型文件预期位于
DeepLIIF/model-server/DeepLIIF_Latest_Model。 - 默认情况下,序列化文件将保存到与输入模型文件相同的目录。
测试
要测试模型:
deepliif test --input-dir /path/to/input/images
--output-dir /path/to/output/images
--model-dir /path/to/the/serialized/model
--tile-size 512
或
python test.py --dataroot /path/to/input/images
--results_dir /path/to/output/images
--checkpoints_dir /path/to/model/files
--name Model_Name
- 预训练模型的最新版本可以在这里下载。
- 在对图像进行测试之前,必须按上述方法序列化模型文件。
- 序列化的模型文件预期位于
DeepLIIF/model-server/DeepLIIF_Latest_Model。 - 测试结果将保存到指定的输出目录,默认为输入目录。
- 必须指定瓦片大小,用于将图像分割成瓦片进行处理。瓦片大小基于输入图像的分辨率(扫描放大倍数),建议的值是40倍图像使用512的瓦片大小,20倍使用256,10倍使用128。注意,瓦片大小越小,推理所需时间越长。
- 测试数据集可以在这里下载。
全幻灯片图像(WSI)推理:
对于全幻灯片图像的转换和分割,
你可以简单地使用相同的测试命令,
将包含全幻灯片图像的目录路径作为input-dir。
DeepLIIF自动逐区域读取WSI,
分别转换和分割每个区域,然后拼接这些区域
以创建整个幻灯片图像的转换和分割结果,
然后以ome.tiff格式将所有掩码保存在给定的output-dir中。
根据可用的GPU资源,可以更改region-size。
deepliif test --input-dir /path/to/input/images
--output-dir /path/to/output/images
--model-dir /path/to/the/serialized/model
--tile-size 512
--region-size 20000
如果您愿意,可以使用Torchserve运行模型。 请参阅文档了解如何使用Torchserve部署模型以及运行推理的示例。
Docker
我们提供了一个Dockerfile,可用于在容器内运行DeepLIIF模型。 首先,您需要安装Docker引擎。 安装Docker后,您需要按照以下步骤操作:
- 在此处下载预训练模型并将其放置在DeepLIIF/model-server/DeepLIIF_Latest_Model中。
- 从docker文件创建docker镜像:
docker build -t cuda/deepliif .
然后将该镜像用作基础。您可以复制并使用它来运行应用程序。应用程序需要一个隔离的环境来运行,称为容器。
- 创建并运行容器:
docker run -it -v `pwd`:`pwd` -w `pwd` cuda/deepliif deepliif test --input-dir Sample_Large_Tissues --tile-size 512
从镜像运行容器时,deepliif CLI将可用。
您可以在激活的环境中轻松运行任何CLI命令,并将结果从docker容器复制到主机。
ImageJ插件
如果您没有GPU或适当的硬件访问权限,只想使用ImageJ运行推理,我们还为您创建了一个ImageJ插件以方便使用。
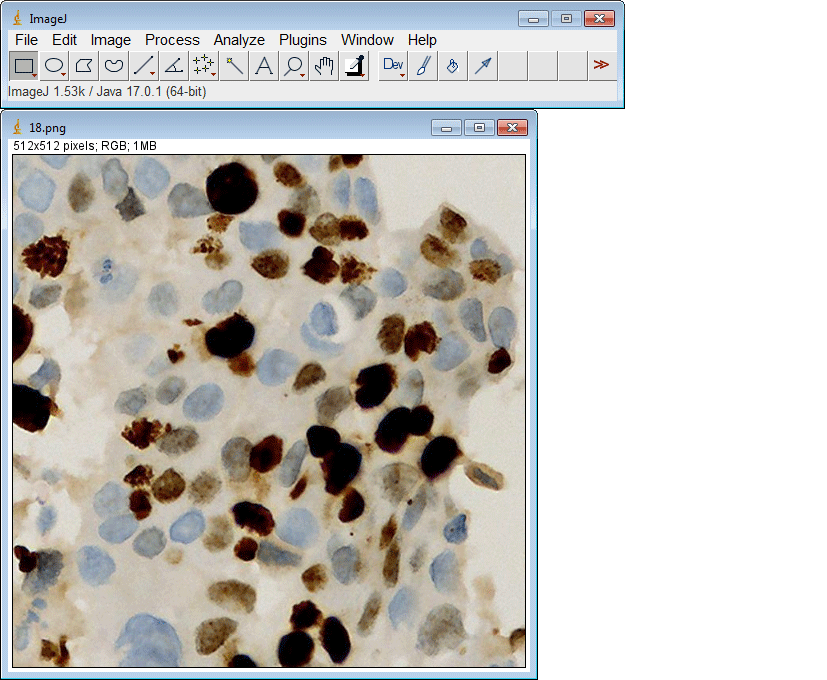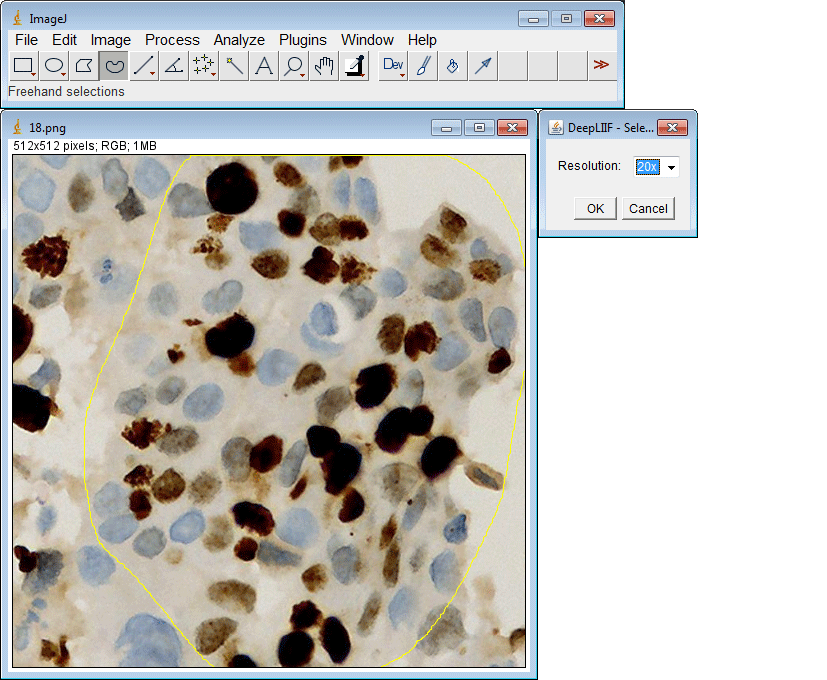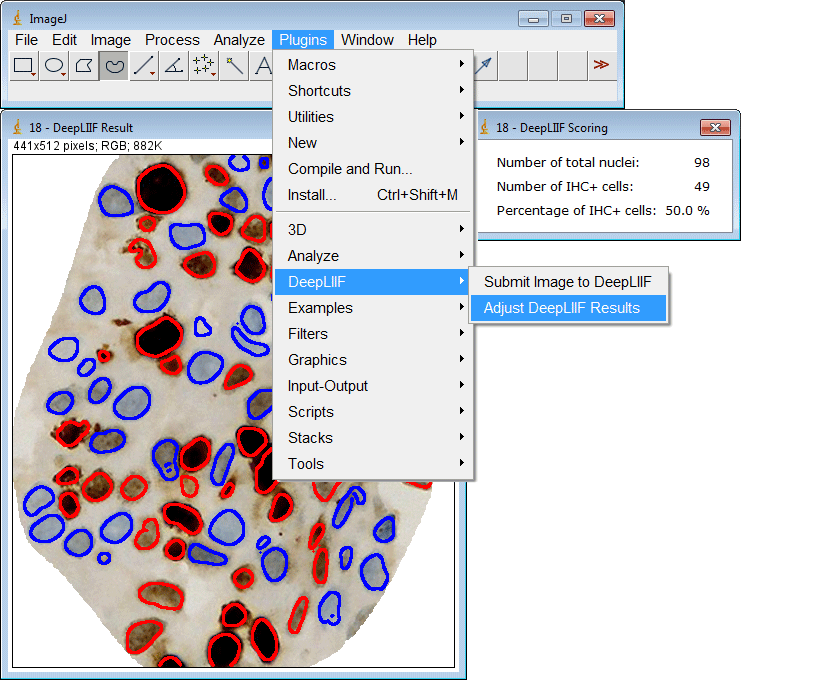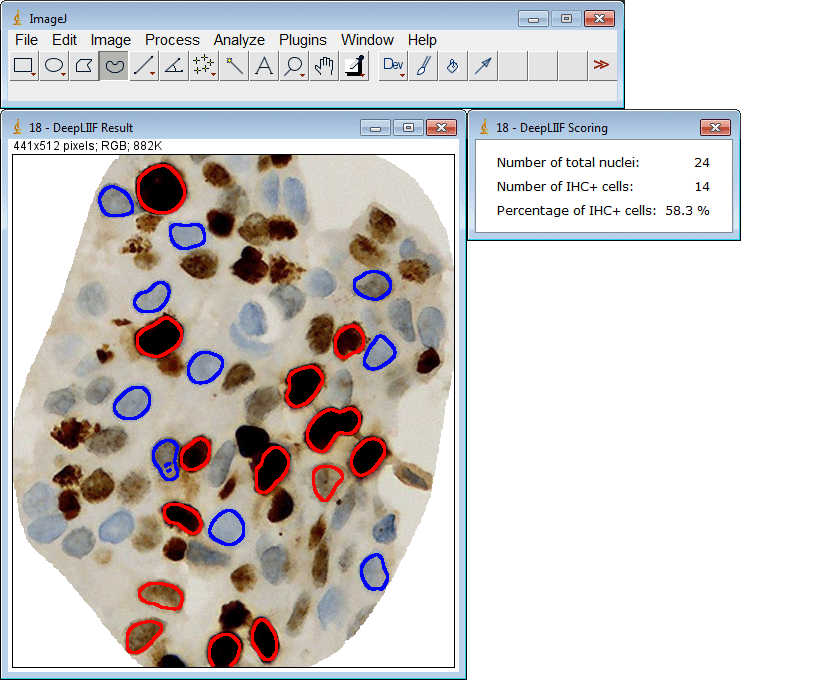
该插件还支持一次提交多个ROI:
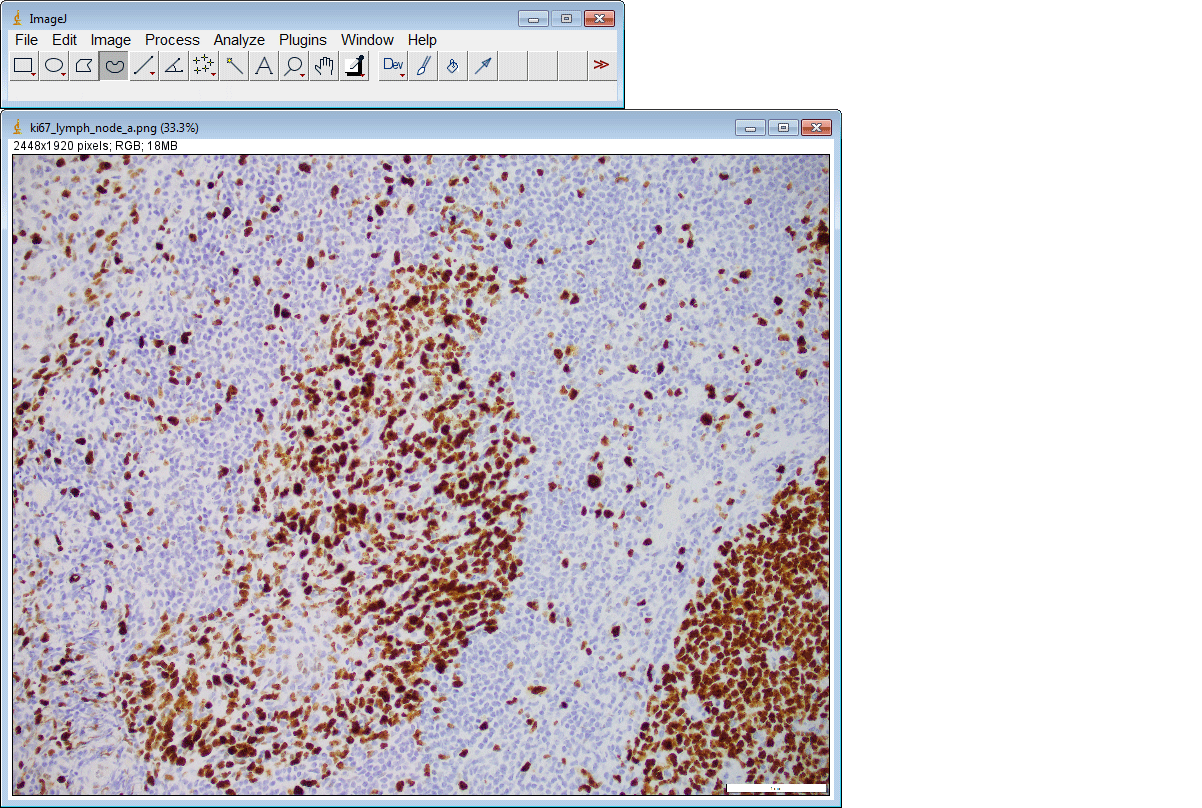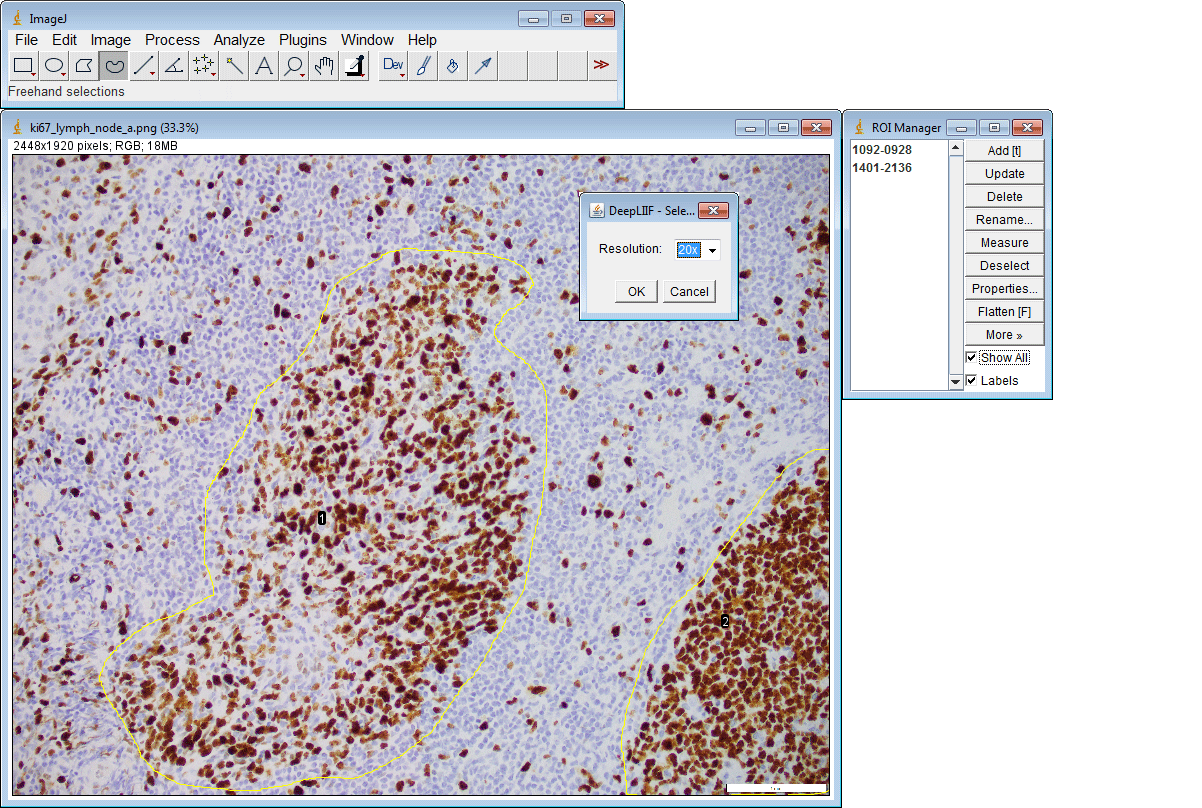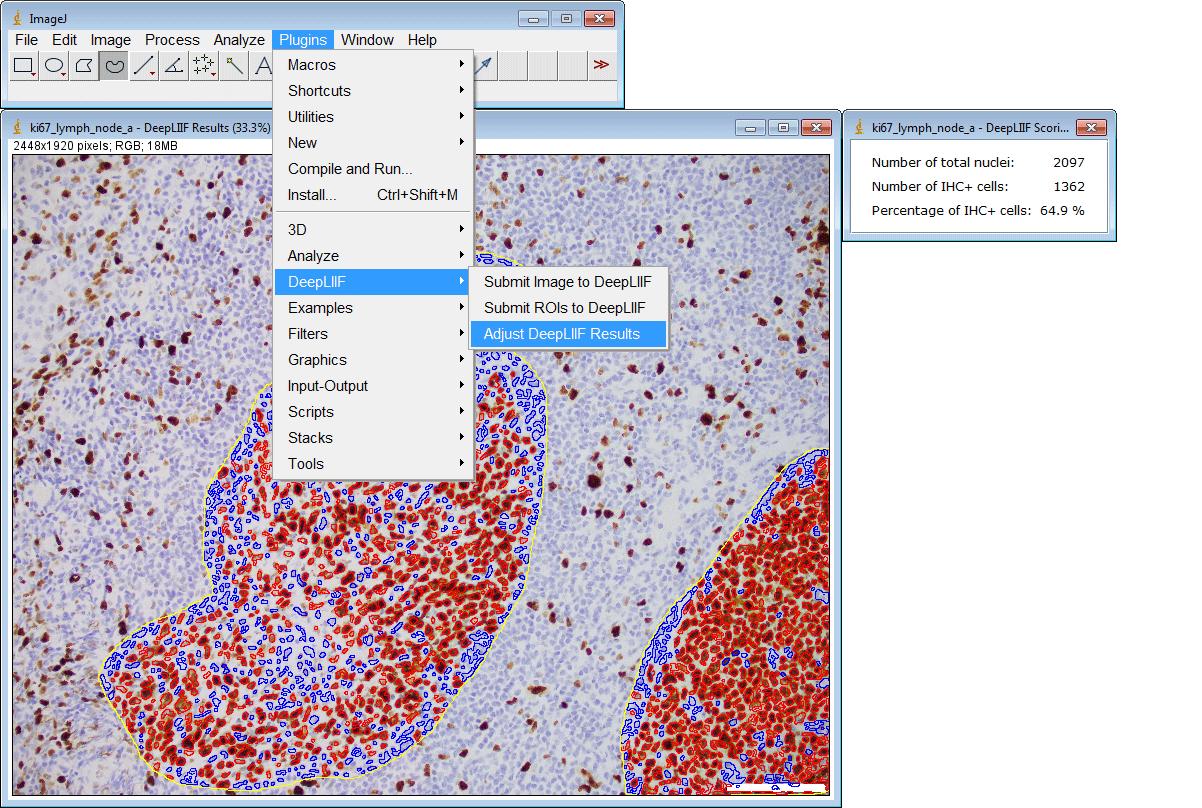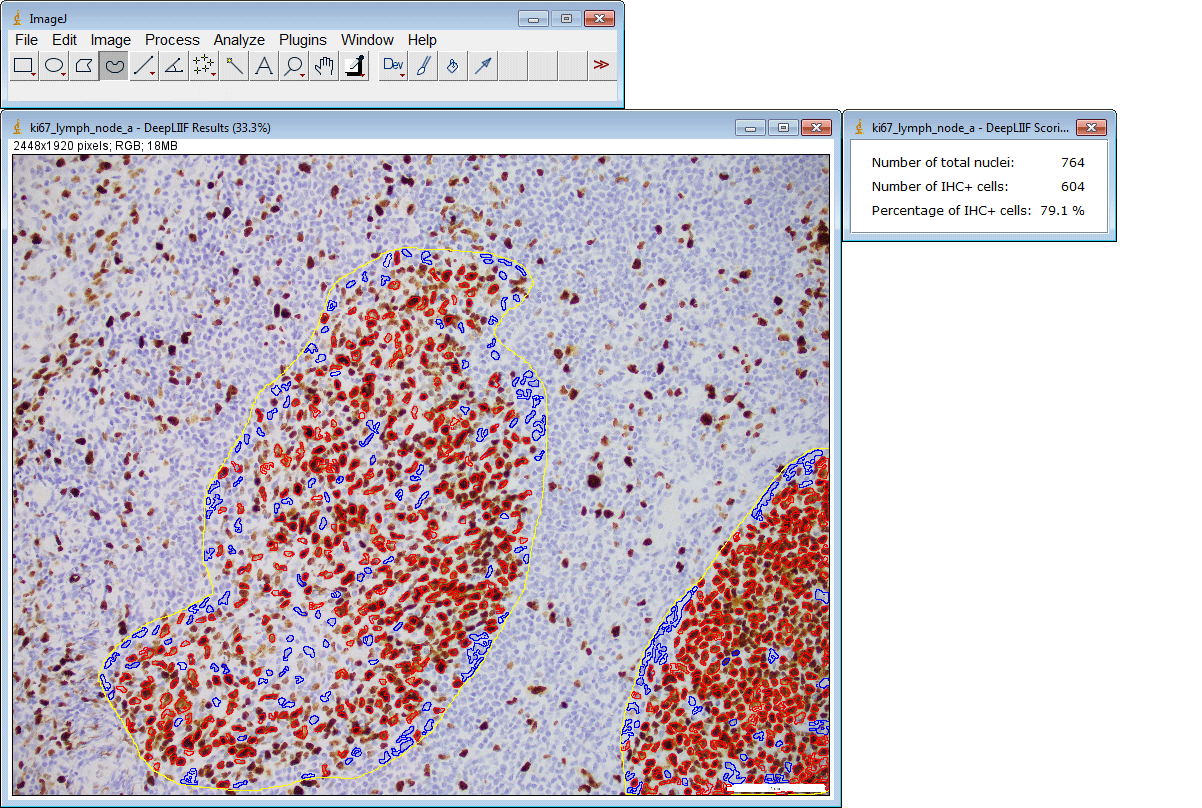
云部署
如果您没有GPU或适当的硬件访问权限,也不想安装ImageJ,我们还创建了一个云原生DeepLIIF部署,具有用户友好的界面,可以上传图像、可视化、交互并下载最终结果。
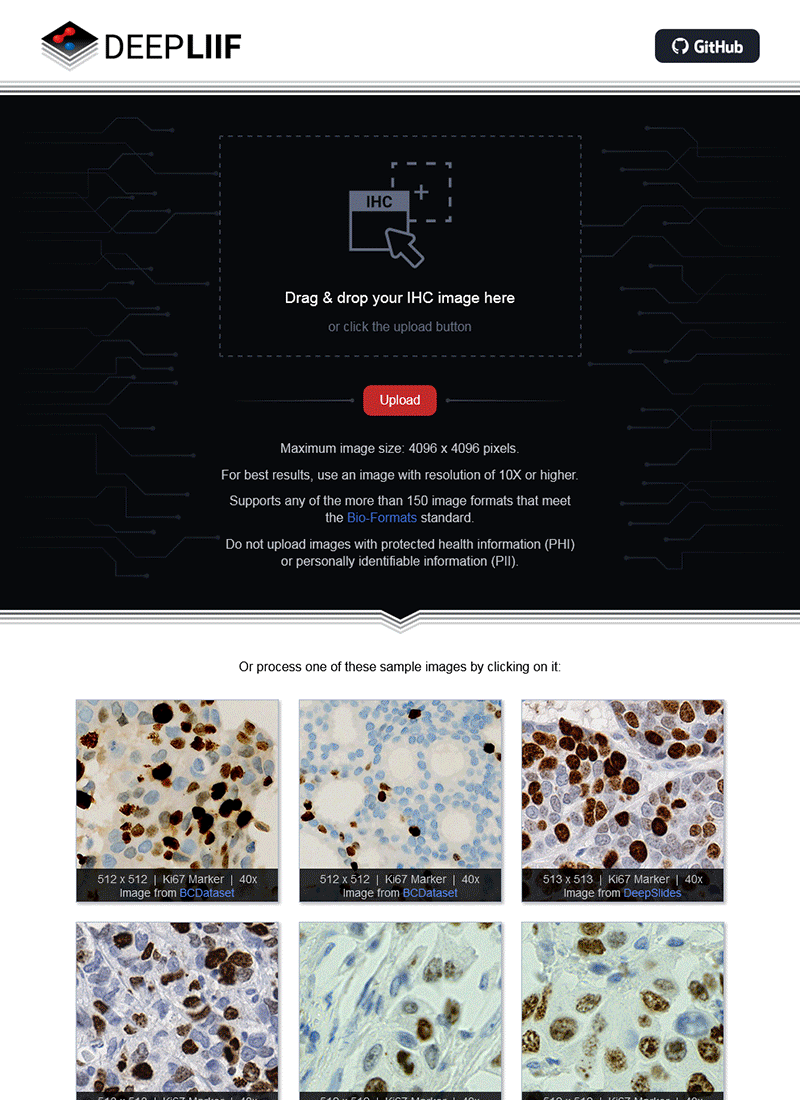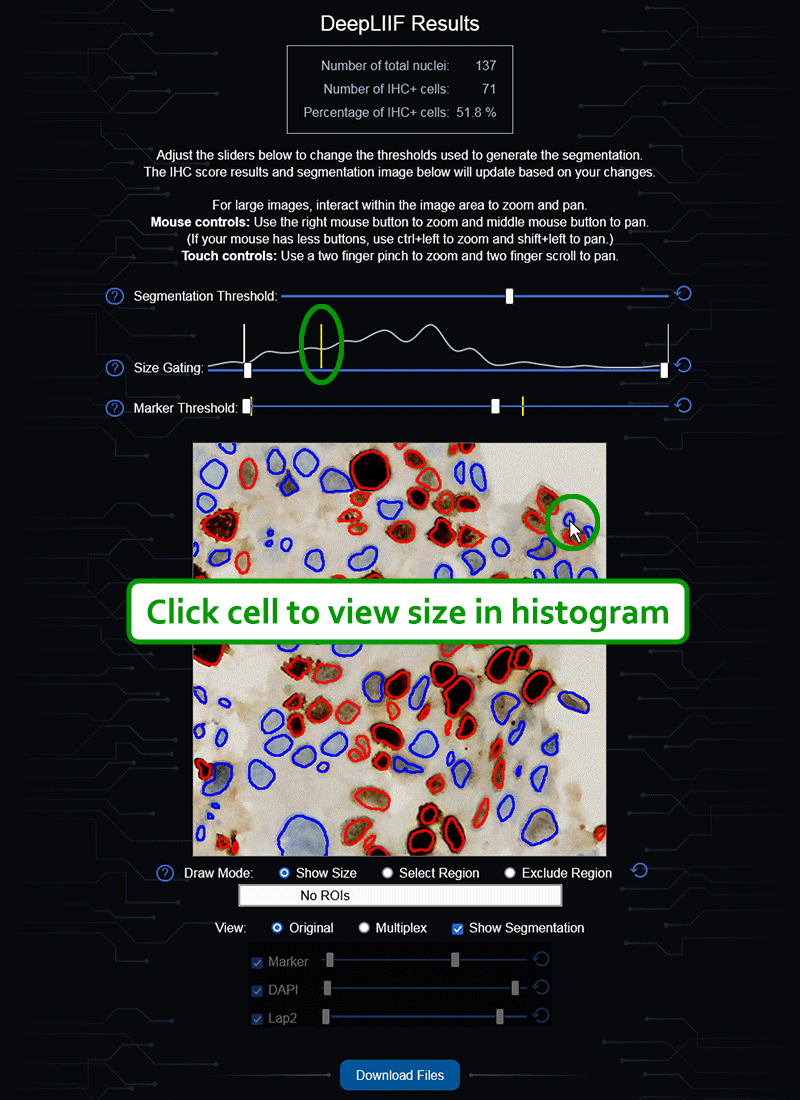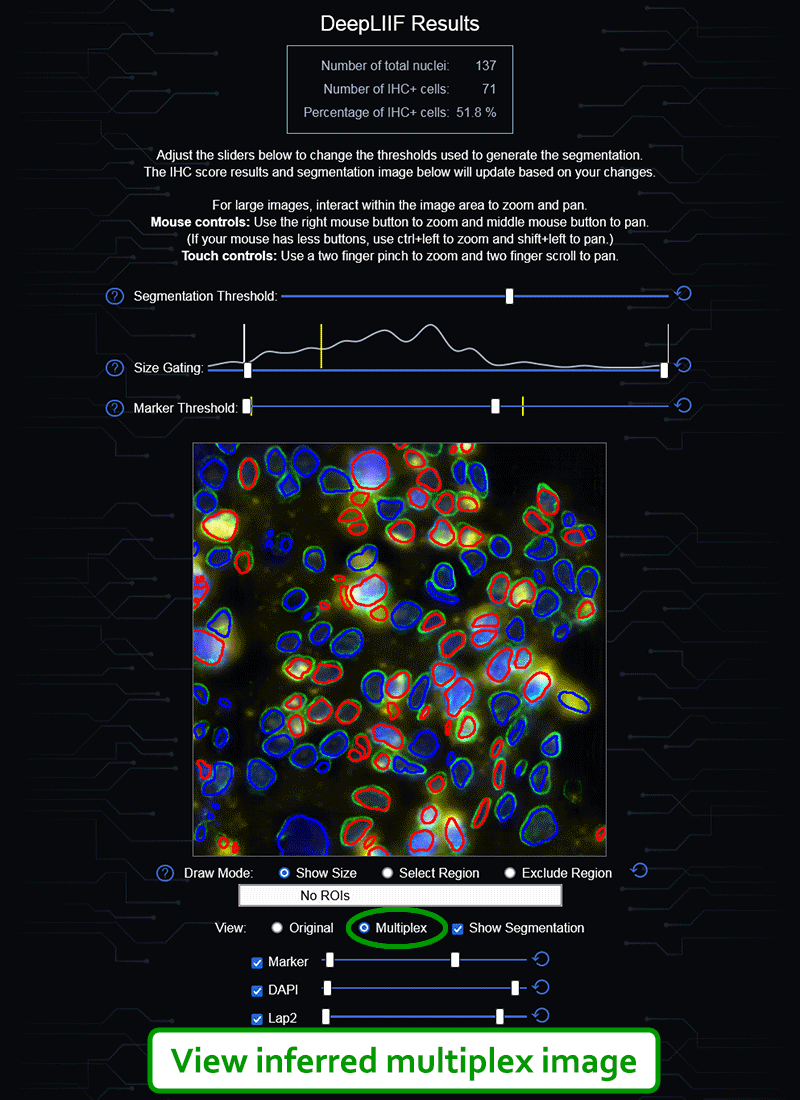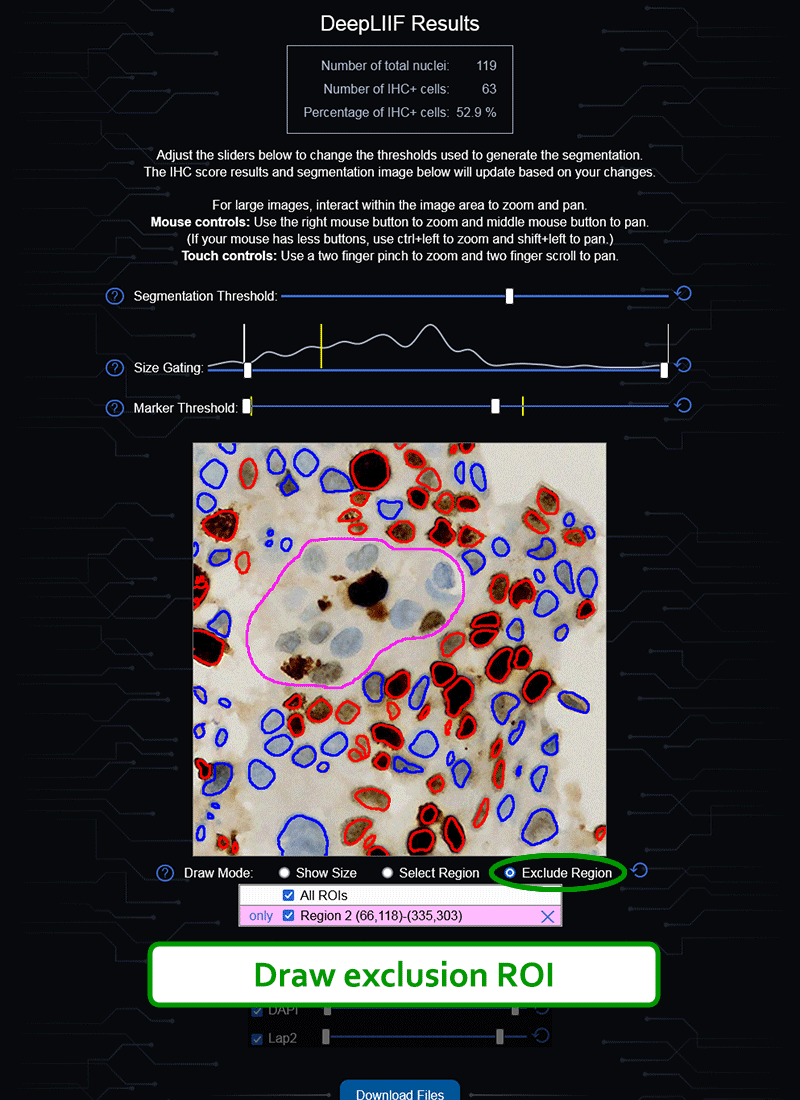
云API端点
DeepLIIF还可以通过端点以编程方式访问,方法是发送包含原始图像文件的多部分编码请求,以及可选参数,包括后处理阈值:
POST /api/infer
文件参数:
img(必需)
要运行DeepLIIF的图像。
查询字符串参数:
resolution
用于扫描幻灯片的分辨率(10x、20x、40x)。默认为40x。
pil
如果存在,使用Pillow而不是Bio-Formats加载图像。Pillow更快,但仅适用于常见图像类型(png、jpeg等)。
slim
如果存在,仅返回优化后的分割结果图像。
nopost
如果存在,不执行后处理(仅返回推理图像)。
prob_thresh
在后处理推断的分割图像时使用的概率阈值。分割图值必须高于此值,才能将像素包含在最终的细胞分割中。有效值为0-254范围内的整数。默认为150。
size_thresh
后处理中细胞大小门控的下限阈值。分割的细胞必须拥有比此值更多的像素,才能包含在最终的细胞分割中。有效值为0、正整数或"auto"。"auto"将尝试根据检测到的细胞大小分布自动确定此下限。默认为"auto"。
size_thresh_upper
后处理中细胞大小门控的上限阈值。分割的细胞必须拥有少于此值的像素,才能包含在最终的细胞分割中。有效值为正整数或"none"。"none"将在大小门控中不使用上限阈值。默认为"none"。
marker_thresh
推断的标记图像对后处理中细胞分类为阳性的影响阈值。如果细胞的任何对应像素在标记图像中高于此阈值,则无论推断的分割图像中的值如何,该细胞都将被分类为阳性。有效值为0-255范围内的整数、"none"或"auto"。"none"将在分类过程中不使用标记图像。"auto"将自动从标记图像确定阈值。默认为"auto"。
例如,在Python中:
import os
import json
import base64
from io import BytesIO
import requests
from PIL import Image
# 使用主DeepLIIF仓库中的样本图像
images_dir = './Sample_Large_Tissues'
filename = 'ROI_1.png'
root = os.path.splitext(filename)[0]
res = requests.post(
url='https://deepliif.org/api/infer',
files={
'img': open(f'{images_dir}/{filename}', 'rb'),
},
params={
'resolution': '40x',
},
)
data = res.json()
def b64_to_pil(b):
return Image.open(BytesIO(base64.b64decode(b.encode())))
对于data['images']中的每个名称和图像:
使用f'{images_dir}/{root}_{name}.png'作为文件名以写入模式打开文件:
将b64_to_pil(img)转换后的图像以PNG格式保存到文件中
使用f'{images_dir}/{root}_scoring.json'作为文件名以写入模式打开文件:
将data['scoring']以JSON格式写入文件,缩进为2
打印data['scoring']的JSON格式,缩进为2
如果你之前已经对一张图像运行过DeepLIIF,想要使用不同的阈值进行后处理,可以直接调用后处理程序,使用先前推断的结果:
POST /api/postprocess
文件参数:
img(必需)
运行DeepLIIF的原始图像。
seg_img(必需)
DeepLIIF先前生成的推断分割图像。
marker_img(可选)
DeepLIIF先前生成的推断标记图像。如果省略此参数,则在分类时不会使用标记图像。
查询字符串参数:
resolution
扫描幻灯片的分辨率(10x、20x、40x)。默认为40x。
pil
如果存在,使用Pillow而不是Bio-Formats加载原始图像。Pillow更快,但仅适用于常见图像类型(png、jpeg等)。Pillow始终用于打开seg_img和marker_img文件。
prob_thresh
用于后处理推断分割图像的概率阈值。分割图的值必须高于此值,像素才能包含在最终的细胞分割中。有效值为0-254范围内的整数。默认为150。
size_thresh
后处理中细胞大小筛选的下限阈值。分割的细胞必须比此值有更多像素才能包含在最终的细胞分割中。有效值为0、正整数或'auto'。'auto'将根据检测到的细胞大小分布自动确定此下限。默认为'auto'。
size_thresh_upper
后处理中细胞大小筛选的上限阈值。分割的细胞必须少于此值的像素才能包含在最终的细胞分割中。有效值为正整数或'none'。'none'表示不使用上限阈值进行大小筛选。默认为'none'。
marker_thresh
推断标记图像在后处理分类中将细胞归类为阳性的影响阈值。如果细胞对应的标记图像中任何像素高于此阈值,该细胞将被归类为阳性,而不考虑推断分割图像的值。有效值为0-255范围内的整数、'none'或'auto'。'none'表示在分类时不使用标记图像。'auto'将自动从标记图像确定阈值。默认为'auto'。(如果未提供marker_img,此参数无效。)
例如,在Python中:
[Python代码示例]
## 合成数据生成
DeepLIIF模型的第一个版本在分离某些大簇中的IHC阳性细胞时存在不足,这是由于我们的训练数据中缺少聚集的阳性细胞。为了向我们的模型注入更多关于聚集阳性细胞的信息,我们提出了一种使用配准数据合成生成IHC图像的新方法。
我们设计了一个基于GAN的模型,该模型接收苏木精通道、mpIF DAPI图像和分割掩码,并生成相应的IHC图像。模型将苏木精通道转换为灰度,以推断更有用的信息(如纹理),并丢弃不必要的信息(如颜色)。苏木精图像指导网络通过保留细胞和背景中伪影的形状和纹理来合成IHC图像的背景。DAPI图像帮助网络识别细胞的位置、形状和纹理,以更好地将细胞与背景分离。分割掩码帮助网络根据细胞类型(阳性细胞:棕色色调,阴性:蓝色色调)指定细胞的颜色。
在下一步中,我们生成具有更多聚集阳性细胞的合成IHC图像。为此,我们通过在分割掩码中选择一定百分比的随机阴性细胞(称为Neg-to-Pos)并将它们转换为阳性细胞来改变分割掩码。下面展示了一些合成IHC图像样本以及原始IHC图像。
*合成IHC图像生成概览。(a) IHC生成器模型的训练样本。(b) 使用训练好的IHC生成器模型合成的一些IHC图像样本。Neg-to-Pos显示了分割掩码中被转换为阳性细胞的阴性细胞百分比。*
我们使用原始IHC图像和合成IHC图像创建了一个新数据集。我们通过将Neg-to-Pos参数设置为50%和70%来对数据集中的每张图像进行两次合成。我们使用新数据集重新训练了我们的网络。您可以在[这里](https://zenodo.org/record/4751737/files/DeepLIIF_Latest_Model.zip?download=1)找到新训练的模型。
## 配准
要对新染色的mpIF和IHC图像进行配准,您可以使用"Registration"目录中的配准框架。请参阅同一目录中提供的README文件了解更多详情。
## 贡献训练数据
为了训练DeepLIIF,我们使用了一个包含肺和膀胱组织的数据集,其中包括使用ZEISS Axioscan扫描的相同组织的IHC、苏木精、mpIF DAPI、mpIF Lap2和mpIF Ki67图像。这些图像经过缩放并使用仿射变换与固定的IHC图像进行配准,最终得到1264组大小为512x512的配准后的IHC和相应多重图像集。我们随机选择了575组用于训练,91组用于验证,598组用于测试模型。我们还从最近发布的[BCDataset](https://sites.google.com/view/bcdataset)中随机选择并手动分割了41张大小为640x640的图像,该数据集包含Ki67染色的乳腺癌切片,并有Ki67+和Ki67-细胞中心点标注(用于细胞检测而非细胞实例分割任务)。我们将这些图块分割成164张大小为512x512的图像;测试集在肿瘤细胞密度和Ki67指数方面变化很大。您可以在[这里](https://zenodo.org/record/4751737#.YKRTS0NKhH4)找到这个数据集。
我们还在创建一个可自配置版本的DeepLIIF,它将接受任何配准后的H&E/IHC和多重图像作为输入,并产生最佳输出。如果您正在生成或已经生成了同一切片的H&E/IHC和多重染色(新染色),并希望贡献该数据用于DeepLIIF,我们可以进行配准、通过[ImPartial](https://github.com/nadeemlab/ImPartial)进行全细胞多重分割、训练DeepLIIF模型,并将其发布回社区,同时充分认可贡献者的贡献。
- [x] **纪念斯隆凯特琳癌症中心** [用于乳腺癌、肺癌和膀胱癌的AI就绪免疫组织化学和多重免疫荧光数据集](https://zenodo.org/record/4751737#.YKRTS0NKhH4)(**Nature Machine Intelligence'22**)
- [x] **莫菲特癌症中心** [用于头颈部鳞状细胞癌的AI就绪多重免疫荧光和多重免疫组织化学数据集](https://wiki.cancerimagingarchive.net/pages/viewpage.action?pageId=70226184)(**MICCAI'23**)
## 支持
请使用[Image.sc论坛](https://forum.image.sc/tag/deepliif)进行与DeepLIIF相关的讨论和问题。
可以在[GitHub问题](https://github.com/nadeemlab/DeepLIIF/issues)标签页报告错误。
## 许可
© [Nadeem实验室](https://nadeemlab.org/) - DeepLIIF代码在**Apache 2.0 with Commons Clause**许可下分发,仅供非商业学术目的使用。
## 致谢
* 本代码受到[PyTorch中的CycleGAN和pix2pix](https://github.com/junyanz/pytorch-CycleGAN-and-pix2pix)的启发。
## 参考
如果您在研究中发现我们的工作有用,或者使用了部分代码或我们发布的数据集,请引用以下论文:
@article{ghahremani2022deep, title={Deep learning-inferred multiplex immunofluorescence for immunohistochemical image quantification}, author={Ghahremani, Parmida and Li, Yanyun and Kaufman, Arie and Vanguri, Rami and Greenwald, Noah and Angelo, Michael and Hollmann, Travis J and Nadeem, Saad}, journal={Nature Machine Intelligence}, volume={4}, number={4}, pages={401--412}, year={2022}, publisher={Nature Publishing Group} }
@article{ghahremani2022deepliifui, title={DeepLIIF: An Online Platform for Quantification of Clinical Pathology Slides}, author={Ghahremani, Parmida and Marino, Joseph and Dodds, Ricardo and Nadeem, Saad}, journal={Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR)}, pages={21399--21405}, year={2022} }
@article{ghahremani2023deepliifdataset, title={An AI-Ready Multiplex Staining Dataset for Reproducible and Accurate Characterization of Tumor Immune Microenvironment}, author={Ghahremani, Parmida and Marino, Joseph and Hernandez-Prera, Juan and V. de la Iglesia, Janis and JC Slebos, Robbert and H. Chung, Christine and Nadeem, Saad}, journal={International Conference on Medical Image Computing and Computer-Assisted Intervention (MICCAI)}, year={2023} }
@article{nadeem2023ki67validationMTC,
作者 = {Nadeem, Saad 和 Hanna, Matthew G 和 Viswanathan, Kartik 和 Marino, Joseph 和 Ahadi, Mahsa 和 Alzumaili, Bayan 和 Bani, Mohamed-Amine 和 Chiarucci, Federico 和 Chou, Angela 和 De Leo, Antonio 和 Fuchs, Talia L 和 Lubin, Daniel J 和 Luxford, Catherine 和 Magliocca, Kelly 和 Martinez, Germán 和 Shi, Qiuying 和 Sidhu, Stan 和 Al Ghuzlan, Abir 和 Gill, Anthony J 和 Tallini, Giovanni 和 Ghossein, Ronald 和 Xu, Bin},
标题 = {髓样甲状腺癌中Ki67增殖指数:多种计数方法的比较研究以及图像分析和深度学习平台的验证},
期刊 = {组织病理学},
年份 = {2023},
doi = {https://doi.org/10.1111/his.15048}
}
@article{zehra2024deepliifstitch,
作者 = {Zehra, Talat 和 Marino, Joseph 和 Wang, Wendy 和 Frantsuzov, Grigoriy 和 Nadeem, Saad},
标题 = {重新思考低资源环境下的组织学切片数字化工作流程},
期刊 = {国际医学图像计算与计算机辅助干预会议(MICCAI)},
年份 = {2024}
}











